核心理念原文链接
dN/dS 是什么
这里的 S 即 Synonymous,N 即 Nonsynonymous。
dN 指的是非同义替换率,dS 指的是同义替换率。但我认为另一个知乎博主文章里的说法会更加直观一些:
- dN: 每个非同义位点上发生的非同义突变的数量。
- dS: 每个同义位点上发生的同义突变的数量。
计算原理
首先,简单来说,dN 和 dS 的计算涉及到四个变量,分别为同义突变数量(S)、非同义突变数量(N)以及同义位点数量(Ssite)和非同义位点数量(Nsite)。
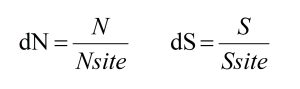
前两个变量 N 和 S 非常好理解,所以这里需要重点关注的是后两个变量 Nsite 和 Ssite,即每种类型的位点数量是什么?它们是如何得到的?但在这之前,我们先简单回顾一下一些常见的突变类型。
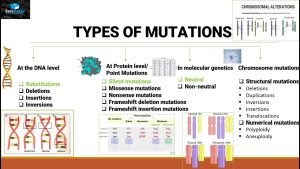
突变类型
我们都知道,一个密码子在发生突变时,其性质可能发生以下几种变化,以密码子 TTT 为例(其编码苯丙氨酸 Phe):
- 错义突变:由编码一个氨基酸变为编码另一个氨基酸,例如 TTT(苯丙氨酸)>ATT(异亮氨酸)。
- 同义突变:突变过后,仍编码相同氨基酸,例如 TTT(苯丙氨酸)>TTC(苯丙氨酸)。同义突变通常发生在密码子的第三个碱基上(有时第一个碱基也可以,例如 AGA>CGA 皆编码精氨酸),此时我们称这个位点具有简并性。需要注意的是,在某些情况下,终止密码子突变为终止密码子(例如 TAA>TAG)也被视为一种同义突变。
除了这一些可能的突变以外,还有一些其他的情况,例如,TAT(酪氨酸) 可能变为一个终止密码子(TAA),这时我们称其为无义突变,在某些情景下,这也可以被称为截断突变 (protein-truncating variant,PTV)。
以上突变类型并不涵盖全部情况,但这些突变是大多 dN/dS 算法的关注对象。
部分情况下,错义突变和无义突变会被统一视为非同义突变,当然在一些算法中,无义突变并不被考虑。这也取决于具体的研究目的和需要。
位点类型
现在我们知道,一个位点的突变可能带来的结果也不同,这也是计算 Ssite 和 Nsite 的关键所在。
首先对于任何情况下的 Nsite 和 Ssite 计算结果,我们都有:
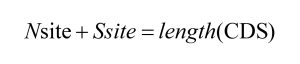
例如,对于一个长度为 3 的密码子,它所计算出来的 Nsite 和 Ssite 总和也必定为 3。
接下来就是计算过程,最简单的计算即遍历每个位点的每种突变情况可能导致的突变类型,对于 TTT,我们可以列举出它的所有可能突变情况,这里直接用杨子恒《计算分子进化》中的一张图作为示例:
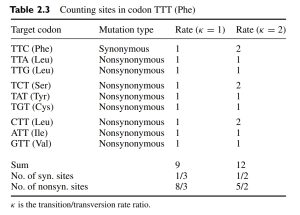
我们先仅考虑 k=1 一栏,k=2 在后文中我们会拓展讨论。
其中,对于一个密码子的每一个位点,它都可以突变成除它自身之外的碱基,因此每个位点都有三个 Target codon,其中同义突变的情况仅有 TTT>TTC,而其他都为非同义突变。
因此,这里共有 9 个情况,其中同义突变占 1 个,非同义突变占 8 个。因此,同义位点数量为 1/9 3(3 即密码子长度),非同义位点数量为 8/9 3。最后则分别为表中的 1/3 和 8/3。
突变计数及 dN/dS 计算
知道位点数量后,我们就可以计算了,依然以一个最简单的情况作为示例,假设我们有一个包含九个 TTT 的 CDS:
-
TTTTTTTTTTTTTTTTTTTTTTTTTTT
通过计算我们可以知道,它具有 3 个同义位点和 24 个非同义位点,假设这里在两个密码子上我们观察到了一个同义突变和一个非同义突变: -
TTCTTTTTTTTTTTTTTTTTTTTTTAT
那么此时,其 dN = 1/24,dS = 1/3,计算得到 dN/dS = 1/8。
当然,在突变计数步骤中也可能出现更加复杂的情况,例如同一个密码子上出现了两个突变,例如 TTT 变为了 TCA,这种情况就要考虑多条进化途径:
- TTT > TTA > TCA(苯丙氨酸 > 亮氨酸 > 丝氨酸),这条途径涉及到两个非同义突变。
- TTT > TCT > TCA(苯丙氨酸 > 丝氨酸 > 丝氨酸),这条途径涉及到一个非同义突变和一个同义突变。
对于这种情况,常见的做法有对不同的途径赋予合适的权重等,更多细节如果看客感兴趣可自行深入了解。
dN/dS 应该是多少
在最简单的 dN/dS 计算中(前文中的 k=1),其中有一个隐性的假设:我们认为每一个位点上每一个突变情况发生的概率都是相等的。
那么,我们继续以 TTT 作为例子,如果每一个突变情况发生的概率都是相等的,理想情况下(无选择压力)我们会获得的 dN/dS 值会是多少?
很简单,发生同义突变的概率是 1/9,发生非同义突变的概率是 8/9,假设发生突变的数量为 x,则通过计算和约分后我们可以发现,dN/dS 值应该是 1。
当然,在现实条件中,每一个突变情况发生的概率并不一定相等,例如,转换(transition)发生的概率就要比颠换(transversion)更高,而很多同义突变都是转换事件,因此如果我们忽略转换和颠换之间的发生概率差异,就会导致 Ssite 低估和 Nsite 的高估,从而导致计算的 dN/dS 偏低。
对于上面这种情况,一种可行的方法就是在计算转换事件时,给它提供的位点数量进行相应加权,也就有了这个表格中 k=2 的情况:
相似的事情还发生在密码子偏好等因素上,而很多方法也针对这些点进行了改进。例如一篇 17 年的 cell 文章中,作者使用 192 个 rate parameters (包括不同的替换类型和链属性,有些类似 3-mer 的突变率模型) 来计算 dN/dS,并发现其他方法要么高估要么低估了 dN/dS。
所以,dN/dS 有一个很关键的设计理念,就是在完全没有选择的情况下,这个值应当等于 1,即同义替代率和非同义替代率应当是相等的。这个概念对于后续的一系列引申也很重要。
dN/dS 有什么用
现在我们能够知道,如果完全考虑了不同替换发生概率的差异并针对它们矫正 Nsite 和 Ssite 的计算,那么 dN/dS 在没有选择的情况下应该是等于 1 的。
在很多地方我们也可以看到这么一个结论:
- dN/dS < 1,表明存在纯化选择。
- dN/dS = 1,表明处在中性选择下。
- dN/dS > 1,表明存在正选择。
也就是说,dN/dS 还可以帮助我们确认一个基因所处的状态,并据此推断出其选择的机制,为我们理解其进化过程提供信息。
那么为什么说 dN/dS = 1 时,基因处于中性选择下呢?这里首先我们要明确一个概念,中性选择并不意味着不存在选择,只是它对生物体的适应性和生存几率没有正面或负面的影响,因此它并不因自然选择而频率增加或下降,在种群内的频率变化全由遗传漂变决定。当然为了方便理解,可以将其视为和 “没有选择” 等效。
同样,中性(neutral)的概念也是如此,一个中性突变就是对生物既无益处也无害处的突变(亦或是带来的益害相抵消)。
然后要明确一个概念,dN/dS = 1 推导出中性选择的前提假设是 —— 同义突变都是中性的。
不妨设想一下,如果同义突变和非同义突变都受到相同强度的强纯化选择,导致其突变数量减少 50%,那么最后算出来的 dN/dS 依然等于 1。
那么这个假设到底成不成立呢?很显然不成立。这个假设主要的根基在于同义突变并不会改变蛋白质的组成和结构,因此从蛋白的角度出发它不会对生物体的表型产生影响。但是在转录翻译阶段,已经有很多研究发现,不同密码子就算编码同一氨基酸,也会存在效率和质量上的不同(这也是密码子偏好的原因之一),所以大多数同义突变其实归根结底是有害的(可见 Shen et al. 2022, Nature )。
但是这影不影响我们使用同义突变作为中性的 Proxy?其实这是个很有争议的问题,因为即使同义突变有害,也很难去量化同义突变到底 “多有害”。今年 4 月份,针对得出 “大多数同义突变都有害” 的文章有人产生了质疑。同样,很多其他的研究表明同义突变可能产生影响的概率比非同义小得多。
违反一个假设的严重性,一要看违反的程度,二要看理论对此的鲁棒性。就以我个人目前进行的分析来看,同义突变受到的影响应该较为轻微。从各种使用 dN/dS 进行选择推断的分析来看,似乎其结果都 “合乎情理”。当然,具体情况如何也有待更详尽的探讨。
原理总结
- 计算 Nsite 和 Ssite 时,对于突变率高的突变类型,需要对其进行一定的加权以平衡其对 dN 和 dS 计算的贡献。以往的计算中,转换-颠换比率 和 密码子偏好 等都是基于该点考虑对计算进行修正。
- 基于突变发生概率矫正后计算的 dN/dS 可以作为选择情况的参考。如果同义突变的中性假设是正确的或者几近正确的,则当 dN/dS 等于 1 时我们可以认为该蛋白编码基因处于中性选择下。
软件计算dN/dS
安装
library(devtools)
install_github("im3sanger/dndscv")下载所需的参考文件(dndscv 默认hg19)
wget https://codeload.github.com/im3sanger/dndscv_data/zip/refs/heads/master加载R包
library(vcfR)
library(dndscv)
library(IRanges)
library(dplyr)vcf 文件处理
vcf <- read.vcfR("/data/shumin/software/snpEff/Somatic/CC56tissueA_snpEff_annotated.vcf")
# 提取位置信息
pos_info <- as.data.frame(getFIX(vcf))
# 提取INFO字段中的ANN信息
ann_info <- extract.info(vcf, element = "ANN")
# 提取必要的列
variants_1 <- data.frame(
chr = pos_info$CHROM,
pos = pos_info$POS,
ref = pos_info$REF,
mut = pos_info$ALT
)
# 解析注释信息条目
parsed_annotations <- lapply(ann_info, function(ann_info) {
# 根据 | 分割注释信息
fields <- unlist(strsplit(ann_info, "|", fixed = TRUE))
# 返回需要的字段,例如基因名和变异类型
gene_name <- fields[4] # 基因名在第四个位置
variant_type <- fields[2] # 变异类型在第二个位置
impact <- fields[3] # 影响程度通常在第三个位置
return(list(Gene = gene_name, Variant_Type = variant_type, Impact = impact))
})
# 将列表转换为数据框
variants_2 <- do.call(rbind, parsed_annotations)
# 合并数据
variants <- cbind(variants_1, variants_2)
# 将属性为list的列转换为字符串
variants$Gene <- sapply(variants$Gene, function(x) paste(x, collapse = ""))
variants$Variant_Type <- sapply(variants$Variant_Type, function(x) paste(x, collapse = ", "))
variants$Impact <- sapply(variants$Impact, function(x) paste(x, collapse = ""))
# 处理缺失值:这里假设直接删除含有NA值的行
variants <- na.omit(variants)
# 确保每列的数据类型正确
variants$chr <- as.character(variants$chr) # 染色体编号
variants$pos <- as.integer(variants$pos) # 位置,转换为整数类型
variants$ref <- as.character(variants$ref) # 参考碱基
variants$mut <- as.character(variants$mut) # 突变碱基
variants$SampleID <- "CC56tissue" # 添加样本名一列
# 移除 "chr" 前缀
variants$chr <- gsub("^chr", "", variants$chr)
# 过滤出单碱基突变(SNPs)
variants <- variants %>%
filter(nchar(ref) == 1 & nchar(mut) == 1)
str(variants)
## 'data.frame': 1624817 obs. of 8 variables:
## $ chr : chr "1" "1" "1" "1" ...
## $ pos : int 13273 14653 14677 14717 16495 16875 17365 17538 17588 17614 ...
## $ ref : chr "G" "C" "G" "G" ...
## $ mut : chr "C" "T" "A" "A" ...
## $ Gene : chr "WASH7P" "DDX11L1" "DDX11L1" "DDX11L1" ...
## $ Variant_Type: chr "downstream_gene_variant" "downstream_gene_variant" "downstream_gene_variant" "downstream_gene_variant" ...
## $ Impact : chr "MODIFIER" "MODIFIER" "MODIFIER" "MODIFIER" ...
## $ SampleID : chr "CC56tissue" "CC56tissue" "CC56tissue" "CC56tissue" ...
# 筛选影响程度“HIGH"的数据进行后续分析
variants_sel <- subset(x=variants, variants$Impact==c('HIGH')) %>% .[,c(8,1:4)]运行dndscv
dndsout <- dndscv(variants_sel, refdb="/usr/local/lib/R/library/dndscv/data/RefCDS_human_GRCh38_GencodeV18_recommended.rda", cv=NULL)结果解读
-
全局结果($globaldnds):这个表包含了所有基因全局的dN/dS值,用于评估正选择、负选择或中性进化
- name: 选择模式的名称(如wmis、wnon、wspl、wtru、wall)。
- mle: 最大似然估计(maximum likelihood estimate)的dN/dS值。
- cilow: dN/dS值的下限95%置信区间。
- cihigh: dN/dS值的上限95%置信区间
dndsout[["globaldnds"]] ## name mle cilow cihigh ## wmis wmis 0.6447645 0.3704856 1.122098 ## wnon wnon 32.2041125 18.5571570 55.887055 ## wspl wspl 17.5535865 9.1980014 33.499495 ## wtru wtru 26.4194841 15.9302538 43.815318 ## wall wall 3.4408418 2.1853716 5.417565
-
基因结果($sel_cv):这个表显示了每个基因的选择信号,用于确定哪些基因可能在癌症中起作用
- gene_name: 基因的名称。
- n_syn: 同义突变的数量。
- n_mis: 错义突变的数量。
- n_non: 无义突变的数量。
- n_spl: 剪接位点突变的数量。
- n_ind: 插入/缺失突变的数量。
- wmis_cv: 错义突变的dN/dS比值(最大似然估计)。
- wnon_cv: 无义突变的dN/dS比值(最大似然估计)。
- wspl_cv: 剪接位点突变的dN/dS比值(最大似然估计)。
- pmis_cv: 错义突变的P值,表示该基因中错义突变的选择压力的显著性。
- ptrunc_cv: 截断突变(无义和剪接位点突变)的P值,表示该基因中截断突变的选择压力的显著性。
- pallsubs_cv: 所有非同义突变(错义、无义和剪接位点突变)的P值,表示该基因中所有非同义突变的选择压力的显著性。
- qmis_cv: 错义突变的q值(FDR校正后的P值)。
- qtrunc_cv: 截断突变(无义和剪接位点突变)的q值(FDR校正后的P值)。
- qallsubs_cv: 所有非同义突变(错义、无义和剪接位点突变)的q值(FDR校正后的P值)。
dndsout[["sel_cv"]] ## gene_name n_syn n_mis n_non n_spl wmis_cv wnon_cv wspl_cv pmis_cv ptrunc_cv pallsubs_cv qmis_cv qtrunc_cv qallsubs_cv ## 15213 SLC9B1 0 0 3 0 0 10531.6440 10531.6440 0.9443649 2.092424e-10 1.611936e-09 0.9855767 4.020802e-06 3.097496e-05 ## 17932 USP17L22 0 0 2 0 0 9729.2562 9729.2562 0.9446037 6.287852e-08 4.273784e-07 0.9855767 5.973446e-04 3.980201e-03 ## 11565 PABPC3 0 0 2 0 0 8019.2737 8019.2737 0.9377189 9.325738e-08 6.213885e-07 0.9855767 5.973446e-04 3.980201e-03 ## 18196 WDPCP 0 0 2 0 0 3805.0805 3805.0805 0.9349721 4.351865e-07 2.744960e-06 0.9855767 2.090636e-03 1.318679e-02 ## 9776 MROH2A 0 0 1 1 0 1761.9124 1761.9124 0.8956018 2.096382e-06 1.176643e-05 0.9855767 8.056814e-03 4.522073e-02 ## 8301 KMT2C 0 0 1 1 0 716.5142 716.5142 0.8386098 1.257247e-05 5.867674e-05 0.9855767 2.906275e-02 1.557393e-01 ## ......
-
突变注释($annotmuts):这个表包含了所有输入的突变及其注释信息
- sampleID: 样本的ID。
- chr: 突变所在的染色体。
- pos: 突变的位置。
- ref: 参考碱基。
- mut: 突变碱基。
- gene: 突变所在的基因。
- ref_cod: 参考序列的三联密码子(codon)。
- mut_cod: 突变后的三联密码子(codon)。
- strand: 基因所在的链(正链或负链)。
- ref3_cod: 参考序列的三联密码子(codon)及其上下游核苷酸序列。
- mut3_cod: 突变后的三联密码子(codon)及其上下游核苷酸序列。
- aachange: 氨基酸变化(从参考氨基酸到突变后的氨基酸)。
- ntchange: 核苷酸变化(从参考核苷酸到突变后的核苷酸)。
- impact: 突变的影响(如synonymous、missense、nonsense等)。
dndsout[["annotmuts"]] ## sampleID chr pos ref mut geneind gene ref_cod mut_cod strand ref3_cod mut3_cod aachange ntchange impact pid ## 204 CC56tissue 1 924949 G A 14271 SAMD11 G A 1 GGT GAT - - Essential_Splice ENSP00000480678 ## 10550 CC56tissue 1 12716215 C A 12 AADACL3 C A 1 GCG GAG C13* C39A Nonsense ENSP00000352268 ## 12672 CC56tissue 1 15626753 C T 4093 DDI2 C T 1 ACG ATG R75* C223T Nonsense ENSP00000417748 ## 22914 CC56tissue 1 26553429 T C 14086 RPS6KA1 T C 1 CTG CCG A169A T507C Synonymous ENSP00000363283 ## 34888 CC56tissue 1 44941527 G A 4826 EIF2B3 C T -1 TCA TTA Q145* C433T Nonsense ENSP00000353575 ## 35452 CC56tissue 1 46032703 A G 9269 MAST2 A G 1 TAG TGG V1031V A3093G Synonymous ENSP00000501318 ## ......
-
局部中性测试($sel_loc):包含了局部选择分析的结果,用于识别基因中特定位置的突变是否显示出显著的选择信号。这在识别可能具有功能性影响的热点区域时非常有用
dndsout[["sel_loc"]] ## gene_name n_syn n_mis n_non n_spl wmis_loc wnon_loc wspl_loc pmis_loc pall_loc qmis_loc qall_loc ## 15213 SLC9B1 0 0 8 0 0 10000 0 0.9999991 8.466339e-12 0.9999998 1.626892e-07 ## 11565 PABPC3 0 0 6 0 0 10000 0 0.9999989 5.799150e-08 0.9999998 5.571824e-04 ## 2129 CALHM4 0 0 0 3 0 0 10000 0.9999993 1.035322e-06 0.9999998 6.631585e-03 ## 6152 GCNT7 0 0 0 3 0 0 10000 0.9999991 7.769189e-06 0.9999998 3.732318e-02 ## 18819 ZNF419 0 0 0 3 0 0 10000 0.9999991 3.321557e-05 0.9999998 1.276541e-01 ## 17350 TRIM51GP 0 0 0 3 0 0 10000 0.9999991 7.640284e-05 0.9999998 1.503908e-01 ## ...... signif_genes_localmodel = as.vector(dndsout$sel_loc$gene_name[dndsout$sel_loc$qall_loc<0.1]) print(signif_genes_localmodel) ## [1] "SLC9B1" "PABPC3" "CALHM4" "GCNT7" ## 该模型检测出SLC9B1, PABPC3, CALHM4, GCNT7有显著突变。